EXOSTOSIS HEREDITARIA MÚLTIPLE
RESUMEN DE HISTORIA CLÍNICA:
Paciente masculino de 17 años de edad, que refiere iniciar su padecimiento hace 3 años, con dolor ocasional a nivel de ambas rodillas y tobillos así como discreto aumento de volumen en dichos sitios, acude revisión médica, en donde se le comenta que es debido al crecimiento, por lo que decide iniciar tratamiento analgésico en caso de dolor, con lo que presenta mejoría de los síntomas, sin embargo hace 6 meses nota nuevamente la presencia de dolor y un aumento de volumen de predominio en la rodilla y pierna izquierda así como la palpación de una masa, por lo que vuelve a acudir para valoración médica quien posterior a revisión física le comenta que le palpa una tumoración por lo que se le solicita una tomografía contrastada de miembros pélvicos.
Sin antecedentes heredo-familiares y personales patológicos de importancia.
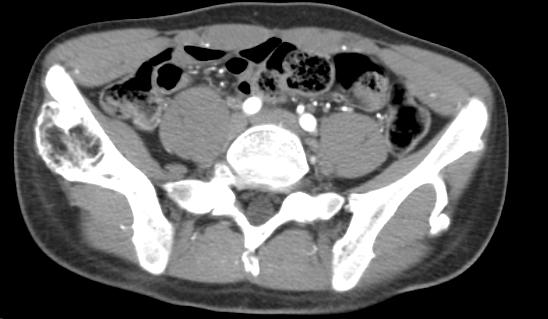
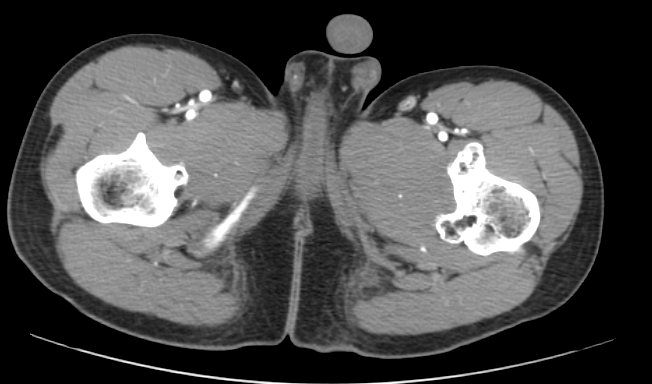
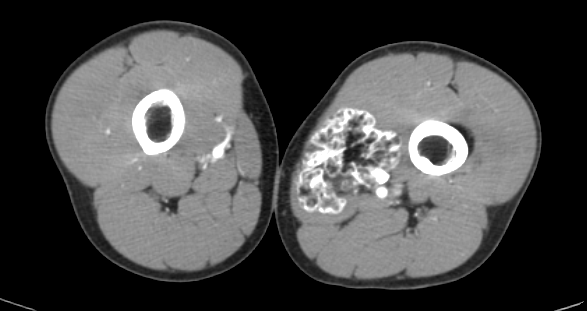
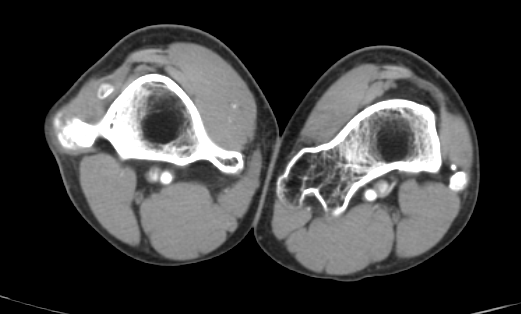

1.- ¿Cuál es el diagnóstico de presunción del caso clínico?
- A. Enfermedad de Ollier.
- B. Metacondromatosis.
- C. Displasia epifisaria hemimélica.
- D. Exostosis hereditaria múltiple.
2.- ¿Característica frecuente de dicha enfermedad?
- A. Múltiples encondromas en las regiones metafisarias.
- B. Continuidad con la corteza ósea y canal medular subyacente.
- C. Cesa su crecimiento cuando las placas de crecimiento se cierran.
- D. B y C son correctas.
3.- ¿Cuál es la frecuencia con la que las lesiones presentan degeneración condrosarcomatosa?
- A. 40%.
- B. 30%.
- C. 5%
- D. 20%.
4.- ¿Complicación más frecuente presentada por este grupo de pacientes?
- A. Fracturas.
- B. Compromiso vascular.
- C. Bursitis
- D. Todas las anteriores.
5.- ¿Hallazgo radiográfico frecuente encontrado?
- A. Matraz de “Erlenmeyer”.
- B. Deformidad de “pseudo-Madelung”.
- C. Lesión intra-articular calcificada.
- D. A y B son correctas.
REVISION BIBLIOGRAFICA:
El osteocondroma representa el tumor óseo más común, es una lesión dependiente del desarrollo en lugar de una verdadera neoplasia, se localizan cerca de las metáfisis de los huesos largos (fémur proximal/distal, húmero proximal, tibia proximal y radio distal). Constituye 20-25% de todos los tumores óseo benignos y el 10-15% de todos los tumores óseos [1].
Compuestos de hueso cortical y medular con cubierta de cartílago hialino suprayacente, y deben demostrar continuidad con la corteza ósea y canal medular subyacente, pueden ser solitarios o múltiples (dos o más osteocondromas), este último asociado con síndrome autosómico dominante, llamado exostosis hereditaria múltiple, osteocondromatosis múltiple o aclasis diafisaria [1].
Las complicaciones asociadas con los osteocondromas son más frecuentes con la forma múltiple e incluye deformidad ósea y cosmética, fracturas, compromiso vascular (estenosis o formación de seudo-aneruismas), secuelas neurológicas, bursitis (exostosis bursata) y transformación maligna condrosarcomatosa (3-5%) [1].
El crecimiento continuo de la lesión y una capa de cartílago hialino de más de 2 cm de grosor después de la madurez esquelética sugiere una transformación maligna. Por lo que se recomienda que sean sometidos a un seguimiento radiográfico periódico, especialmente aquellos con exostosis en los sitios más frecuentes de degeneración sarcomatosa (pelvis y hombro) [3].
Otros hallazgos que sugieran transformación maligna; superficie de la exostosis irregular o indistinta, regiones focales radio-lucidas dentro de la lesión, masa de tejidos blandos con calcificaciones irregulares [4].
Se suelen presentar en la infancia, la mayoría antes de los 12 años, por masa palpable o deformidad con angulación de hueso. Aumentan de tamaño durante la primera década de la vida y dejan de crecer cuando las placas de crecimiento se cierran durante la pubertad. La distribución suele ser simétrica y bilateral.
Pueden ser pediculados o sésiles de base ancha. Estos últimos cuando son más del 90% se correlacionan con deformidad angular grave [1].
El síntoma clínico más común es la presencia de masas exofíticas indoloras, simples o múltiples, cerca de las articulaciones de los huesos largos, que pueden estar asociadas con deformidades óseas [2].
Un tercio de los pacientes muestra deformidad pseudo-Madelung o mano en “bayoneta” en las extremidades superiores debido al acortamiento del cúbito en relación con el radio, o deformidad tipo matraz de “Erlenmeyer” en las extremidades inferiores por estrechamiento meta-diafisario del fémur [2].
La baja estatura es una característica clínica frecuente (40%), puede ser el resultado del desarrollo de exostosis durante la infancia y pubertad temprana [1].
También se ha informado que la tomografía por emisión de positrones con FDG-F18 es precisa en la detección de la des-diferenciación del cartílago y la transformación maligna. [2].
BIBLIOGRAFIA:
- Murphey M., Choi J., Kransdorf M., Flemming D., Gannon F., Imaging of osteochondroma: variants and complications with radiologic-pathologic correlation. RadioGraphics 2000; 20:1407-1434.
- Kok H., Fitzgerald L., Campbell N., Lyburn I., Munk P., Buckley O., Torreggiani W., Multimodality imaging features of hereditary multiple exostoses-. Br J radiol 2013; 86:20120398.
- Nobre Gomes A., Sampaio Silveria C., Santos Paiva R., Monte Aragäo A., Cavalcante Castro J., Chondrosarcoma in a patiente with multiple osteochondromatosis: a case report and review of the literature. Radiol Bras 2006; 39(6): 449-451.
- Pope T., Bloem H., Beltran J., Dysplasias, Musculoskeletal Imaging, Second Edition. Elsevier Saunders. 2015;1087-1101.